
Phenytoin
Order phenytoin 100 mg fast delivery
However medicine 8 pill generic 100mg phenytoin with mastercard, many of the newer antidepressants have been extensively tested in children with other conditions. For example, some antidepressants are approved 154 Essential Psychopharmacology for the treatment of children with obsessive-compulsive disorder. Thus, the safety of some antidepressants is well established in children even if their efficacy for depression is not. Nevertheless, antidepressant treatment studies in children are in progress, and extensive anecdotal observations suggest that antidepressants, particularly the newer, safer ones (see Chapters 6 and 7), are in fact useful for treating depressed children. Thankfully, this is now providing incentives for doing the research necessary to prove the safety and efficacy of antidepressants to treat depression in children, a long neglected area of psychopharmacology. Mania and mixed mania have not only been greatly underdiagnosed in children in the past but also have been frequently misdiagnosed as attention deficit disorder and hyper activity. Furthermore, bipolar disorder misdiagnosed as attention deficit disorder and treated with stimulants can produce the same chaos and rapid cycling state as antidepressants can in bipolar disorder. Thus, it is important to consider the diagnosis of bipolar disorder in children, especially those unresponsive or apparently worsened by stimulants and those who have a family member with bipolar disorder. These children may need their stimulants and antidepressants discontinued and treatment with mood stabilizers such as valproic acid or lithium initiated. Documentation of the safety and efficacy of antidepressants and mood stabilizers is better for adolescents than for children, although not at the standard for adults. That is unfortunate, because mood disorders often have their onset in adolescence, especially in girls. Not only do mood disorders frequently begin after puberty, but children with onset of a mood disorder prior to puberty often experience an exacerbation in adolescence. Synaptic restructuring dramatically increases after age 6 and throughout adolescence. Such events may explain the dramatic rise in the incidence of the onset of mood disorders, as well as the exacerbation of preexisting mood disorders, during adolescence. Unfortunately, mood disorders are frequently not diagnosed in adolescents, especially if they are associated with delinquent antisocial behavior or drug abuse. This is indeed unfortunate, as the opportunity to stabilize the disorder early in its course and possibly even to prevent adverse long-term outcomes associated with lack of adequate treatment can be lost if mood disorders are not aggressively diagnosed and treated in adolescence. The modern psychopharmacologist should have a high index of suspicion and increased vigilance to the presence of a mood disorder in adolescents, because treatments may well be just as effective in adolescents as they are in adults and perhaps more critical to preserve normal development of the individual. According to the monoamine hypothesis, in the case of depression the neurotransmitter is depleted, causing neurotransmitter deficiency. This accumulation theoretically reverses the prior neurotransmitter deficiency (see. Tricyclic antidepressants exert their antidepressant action by blocking the neurotransmitter reuptake pump, thus causing neurotransmitter to accumulate. This accumulation, according to the monoamine hypothesis, reverses the prior neurotransmitter deficiency (see. The tricyclic antidepressants also increased the monoamine neurotransmitters, resulting in relief from depression due to blockade of the monoamine transport pumps. Although the monoamine hypothesis is obviously an overly simplified notion about depression, it has been very valuable in focusing attention on the three monoamine neurotransmitter systems norepinephrine, dopamine, and serotonin. This has led to a much better understanding of the physiological functioning of these three neurotransmitters and especially of the various mechanisms by which all known antidepressants act to boost neurotransmission at one or more of these three monoamine neurotransmitter systems. Monoaminergic Neurons In order to understand the monoamine hypothesis, it is necessary first to understand the normal physiological functioning of monoaminergic neurons. Monoamine neurotransmitters are synthesized by means of enzymes, which assemble neurotransmitters in the cell body or nerve terminal. These differences can be exploited by drugs so that the transport of one monoamine can be blocked independently of another. More recently, adrenergic receptors have been even further subclassified on the basis of both pharmacologic and molecular differences. On the other hand, alpha 2 receptors are the only presynaptic noradrenergic receptors on noradrenergic neurons. Presynaptic alpha 2 re ceptors are important because both the terminal and the somatodendritic re-ceptors are autoreceptors. They are located either on the axon terminal, where they are called terminal alpha 2 receptors, or at the cell body (soma) and nearby dendrites, where they are called somatodendritic alpha 2 receptors. The principal function of the locus coeruleus is to determine whether attention is being focused on the external environment or on monitoring the internal milieu of the body. It helps to prioritize competing incoming stimuli and fixes attention on just a few of these. Thus, one can either react to a threat from the environment or to signals such as pain coming from the body. Where one is paying attention will determine what one learns and what memories are formed as well. Malfunction of the locus coeruleus is hypothesized to underlie disorders in which mood and cognition intersect, such as depression, anxiety, and disorders of attention and information processing. There are many specific noradrenergic pathways in the brain, each mediating a different physiological function. Different receptors may mediate these differential effects of norepinephrine in frontal cortex, postsynaptic beta 1 receptors for mood. The projection from the locus coeruleus to limbic cortex may regulate emotions, as well as energy, fatigue, and psychomotor agitation or psychomotor retardation. A projection to the cerebellum may regulate motor movements, especially tremor. Norepinephrine from sympathetic neurons leaving the spinal cord to innervate peripheral tissues control heart rate. A plethora of dopamine receptors exist, including at least five pharmacological subtypes and several more molecular isoforms. Dopamine 1, 2, 3, and 4 receptors are all blocked by some atypical antipsychotic drugs, but it is not clear to what extent dopamine 1, 3, or 4 receptors contribute to the clinical properties of these drugs. They provide negative feedback input, or a braking action on the release of dopamine from the presynaptic neuron. Most of the cell bodies for noradrenergic neurons in the brain are located in the brainstem in an area known as the locus coeruleus. This is the headquarters for most of the important noradrenergic pathways mediating behavior and other functions such as cognition, mood, emotions, and movements. Some noradrenergic projections from the locus coeruleus to frontal cortex are thought to be responsible for the regulatory actions of norepinephrine on mood. Beta 1 postsynaptic receptors may be important in transducing noradrenergic signals regulating mood in postsynaptic targets. Other noradrenergic projections from the locus coeruleus to frontal cortex are thought to mediate the effects of norepinephrine on attention, concentration, and other cognitive functions, such as working memory and the speed of information processing. Alpha 2 postsynaptic receptors may be important in transducing postsynaptic signals regulating attention in postsynaptic target neurons. The noradrenergic projection from the locus coeruleus to limbic cortex may mediate emotions, as well as energy, fatigue, and psychomotor agitation or psychomotor retardation. The noradrenergic projection from the locus coeruleus to the cerebellum may mediate motor movements, especially tremor. Noradrenergic innervation of the heart via sympathic neurons leaving the spinal cord regulates cardiovascular function, including heart rate, via beta 1 receptors. Noradrenergic innervation of the urinary tract via sympathetic neurons leaving the spinal cord regulates bladder emptying via alpha 1 receptors. The tryptophan transport pump is distinct from the serotonin transporter (see. Serotonin is then stored in synaptic vesicles, where it stays until released by a neuronal impulse. This organization allows serotonin release to be controlled not only by serotonin but also by norepinephrine, even though the serotonin neuron does not itself release nor epinephrine. They convey messages from the presynaptic serotonergic neuron to the target cell postsynaptically.
Order phenytoin amex
They are able to maintain normogly cemia for a long time treatment canker sore discount phenytoin online, then develop postprandial hyperglycemia, and later develop both postprandial and fasting hyperglycemia (ie, hyperglycemia all the time). Thus, a glucose tolerance test to detect postprandial hyperglycemia would be the most sensitive test for diabetes mellitus but is time consuming and difficult to perform in a clinical practice. If there are no clear symptoms of hyperglycemia, the diagnosis of diabetes must be confirmed on a subsequent day by repeat measurement, repeating the same test for confirmation. By using these tests, patients can be classified into one of three categories: (1) normal, (2) impaired glucose tolerance/impaired fasting glucose (ie, prediabetic), or (3) diabetic. Increased risk for microvascular complications of hyperglycemia is seen at a fasting glucose more than 126 mg/dL or A1C >6. Prevention of acute complications of hyperglycemia (eg, diabetic ketoacidosis or nonketotic hyperosmolar hyperglycemia) or hypoglycemia 2. Prevention of long-term complications of hyperglycemia, for example, microvas cular disease such as retinopathy or nephropathy 3. Prevention of long-term complications of macrovascular disease, for example, cardiovascular or cerebrovascular diseasethe foundation of diabetes therapy is dietary and lifestyle modifications. Exercise and even small amounts of weight loss can lower blood pressure and improve glu cose control. Patients should be given instruction in nutrition and encouraged to change sedentary lifestyles. However, most people with diabetes will eventually require medical therapy, and many patients will eventually require a combination of at least two medications. If patients fail to achieve these goals with initial therapy including lifestyle modification and metformin, therapeutic options include adding a second oral or injectable agent, including insulin, or switching to insulin monotherapy. Other routine care in diabetic patients includes frequent physician visits, at least every 3 to 6 months depending on their glucose control, at least yearly ophthal mologic examinations to screen for retinopathy, and yearly urine screens to detect microalbuminuria. Hemoglobin A1C should be checked at least every 3 to 6 months, depending on the patients glucose control. This test allows the physician to know the general glucose control over the preceding 2 to 3 months. Patients without neuropathy should have a foot examination yearly to detect early neuropathic changes; however, those with neuropathy should be examined every 3 months and be instructed on daily self-examination and prevention of injury. She currently takes metformin 1000 mg twice per day, and her fasting morning glucose runs approximately 170 to 200 mg/dL. She states that she conscientiously follows her diet and that she walks 30 minutes to 1 hour daily. Her daughter reports that, on three occasions in the past 2 weeks, her mother became sweaty, shaky, and confused, which resolved when she was given some orange juice. Which of the following conditions is most likely to be contributing to these episodes By diagnostic criteria, this patient falls into the denition of impaired fast ing glucose. Although she does not yet meet the criteria for diabetes, she is at greater risk for developing diabetes in the future and for macrovascular disease. Intensive lifestyle changes (diet and exercise for 30 minutes per day, 5 days per week) can reduce or delay the development of diabetes. Switching from one class of oral agent to another with similar potency would add no benet. Sulfonylureas have long half-lives and can cause prolonged hypoglycemia in elderly patients as well in those with renal insufficiency. Another method, such as insulin, may be more appropriate in this patient, as well as less-intensive control, aiming for an HbA1C of 8% instead of 7%. Physicians should have a high index of suspicion and screen those patients with risk factors. Lifestyle modification and metformin are the initial therapy for most patients initially diagnosed with type 2 diabetes. The major cause of morbidity and mortality in patients with type 2 diabetes mellitus is macrovascular disease, such as coronary artery disease, stroke, and peripheral vascular disease, so aggressive cardio vascular risk factor reduction is essential. The mother reports the patient has always been healthy and has no significant medical history, but she has lost 20 lb recently without trying and has been complaining of fatigue for 2 or 3 weeks. The patient had attributed the fatigue to sleep disturbance, as recently she has been getting up several times at night to urinate. This morning, the mother found the patient in her room, complaining of abdominal pain, and she had vomited. On examination, the patient is slender, lying on a stretcher with eyes closed, but she is responsive to questions. She is afebrile, and has a heart rate 118 bpm, blood pressure 125/84 mm Hg, with deep and rapid respirations at the rate of 24 breaths per minute. Upon standing, her heart rate rises to 145 bpm, and her blood pressure falls to 110/80 mm Hg. Her funduscopic examination is normal, her oral mucosa is dry, and her neck veins are flat. Her chest is clear to auscultation, and her heart is tachycardic with a regular rhythm and no murmur. Her abdomen is soft with active bowel sounds and mild diffuse tenderness, but no guarding or rebound. Urine drug screen and urine pregnancy test are negative, and urinalysis shows no hematuria or pyuria, but 3+ glucose and 3+ ketones. Chest radiograph is read as normal, and plain film of the abdomen has nonspecific gas pattern but no signs of obstruction. She is hypovolemic as a result of osmotic diuresis and has an anion gap metabolic acidosis, which is primarily caused by ketoacids. Her mental status and abdominal pain probably are manifestations of the metabolic acidosis and hyperosmolarity. Careful management and close monitoring will be required to correct fluid and electrolyte deficits and to prevent complications such as hypokalemia and cerebral edema. It is a medical emergency, with an overall mortality rate less than 5% if patients receive prompt and appropriate medical treatment. The majority of episodes are preventable, and many of the deaths also are preventable with proper attention to detail during management. Pathophysiology In the normal physiologic state, there is a fine balance between anabolic and catabolic hormones. This results in storage of energy reserves in the form of triglycerides and glycogen. In the fasting state, insulin serves to inhibit lipolysis, ketogenesis, gluconeogen esis, glycogenolysis, and proteolysis. These effects are critical in controlling the rate of breakdown of energy stores under the influence of catabolic hormones. In the fasting state, it maintains normal glucose levels by stimulating hepatic gluconeogenesis and glycogenolysis. When there is a severe insulin deficiency and a relative excess of glucagon, lipolysis is enhanced, causing release of free fatty acids. Oxidation of the fatty acids produces ketones, such as acetoacetate and beta-hydroxybutyrate, which are organic acids and often referred to as ketoacids. In addition, hyperglycemia produces an osmotic diuresis, which causes severe volume depletion, and electrolyte deficiencies by washing extracel lular sodium, potassium, magnesium, phosphate, and water out of the body. Clinical Presentation Patients with diabetes have an underlying impairment in glucose metabolism and, when challenged by a stress, an increase in insulin requirements. The most common precipi tating events are infections such as pneumonia or urinary tract infection, vascular disorders such as myocardial infarction, or other stressors such as trauma. Diabetic ketoacidosis may be the presentation of new-onset diabetes, or it can occur in patients with established diabetes because of failure to use insulin for whatever rea son or because of use of other medications (eg, glucocorticoids) that interfere with insulin action. Polyuria, polydipsia, weight loss, visual blurring, and decreased mental status are related to hyperglycemia and osmotic diuresis. Nausea, vomiting, abdominal pain, fatigue, malaise, and shortness of breath may be related to the acidosis. Typical signs include reduced skin elasticity, dry mucous membranes, hypoten sion, and tachycardia related to volume depletion.
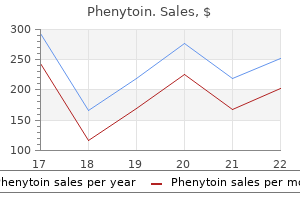
100 mg phenytoin sale
The severity of water deficit is estimated from the plasma electrolytes and body weight medicine zofran generic phenytoin 100mg fast delivery. Thus minimal water loss in adults of normal size addition of solute to the extracellular compartment (causing is approximately 1170 mL/day (670 mL urine plus 500 mL water to move out of cells) and the net loss of water from the insensible loss). A hypernatremia is excessive water loss with inadequate replace history of polyuria and nocturia is important in establishing ment. Some patients are unable to concentrate urine maxi the cause of hypernatremia as diabetes insipidus. Addition of large quanti concentration in normal subjects is 1200 mOsm/L but ties of solute is a rare cause. Volume overload may ics such as furosemide; and use of drugs such as demeclocy cause pulmonary and peripheral edema in these patients. Hypernatremia with polyuria and isos or who have had increased normal losses of water from thenuric urine suggests solute diuresis. Thus, for example, if urine vol makes the diagnosis of hypernatremia, and this will be accom ume is initially 600 mL/h, a 60-kg patient could be expected to panied by plasma osmolality greater than 300 mOsm/kg. Five units of aqueous vasopressin is admin excrete small amounts of concentrated urine. Urine osmolal istered at the end of the test if urine concentration fails to rise. Solute diuresis can be further subdi vided into electrolyte diuresis or nonelectrolyte diuresis. Note that this is the amount of water needed to correct nonelectrolyte solute is the cause of the diuresis. In practice, the patients normal body diuresis is seen with administration of diuretics and is the nor weight may not be known, but only the current body weight. Cerebral edema with neurologic complications has hypernatremia, suggesting diabetes insipidus. A water deprivation test may be neces cells generate and take up idiogenic osmoles, sometimes sary. In this test, a patient with normal or near-normal plasma called organic osmolytes. Since cell volume is determined from osmolality is deprived of water for a scheduled interval while the amount of solute contained within the cell, organic weight, plasma sodium and osmolality, and urine volume and osmolytes resist the movement of water out of the cells and osmolality are measured. Many of the organic centration fails to increase into an appropriately high range osmolytes are taken up from the extracellular space by the (>800 mOsm/kg) despite a plasma osmolality greater than formation of specific membrane channels. For safety when designing the water depri the body theoretically may cause overexpansion of these cells, vation test, patients should be anticipated to continue to maintain resulting in cerebral edema. For example, if [Na+] = 170 meq/L correction may not be necessary in patients who develop for a normally 70-kg patient, 1 L of 0. Therefore, if more rapid anticipated changes in plasma [Na+] in the hypernatremic correction of hypernatremia is desired, hypotonic fluid (5% patient given intravenous fluids. This formula estimates the practice, volume repletion is generally a higher priority, but amount of change in plasma [Na+] when 1 L of any fluid is after some correction of the volume deficit, the water deficit administered: should be addressed directly. This formula demonstrates how little the + + diabetes insipidus, urine volume can exceed 500 mL/h, and plasma [Na ] changes when normal saline ([Na ] = 154 with a severe water deficit, water may have to be given at rates meq/L) is given to a hypernatremic patient. The dose should be adjusted ments of plasma [Na ] are essential because the formula + on the basis of plasma [Na ], urine output, and urine osmo does not account for other fluid sources, urinary losses, or lality. Administration of enough water to maintain will increase the amount of water that must be given. If a reversible patients have renal insufficiency, removal of solute may cause such as lithium toxicity is found, the offending agent require hemodialysis or ultrafiltration with replacement of can be discontinued, although the effect on renal concen water. A few patients have been treated by hemodialysis with trating ability may persist for days. Thiazide diuretics dialysate containing hypotonic solution, facilitating water induce mild volume depletion, leading to increased proxi replacement. Peritoneal dialysis using hypotonic solutions mal tubular sodium reabsorption and decreased delivery of should be efficacious in removing extracellular solute and sodium and water to the distal diluting segment, so that less increasing water replenishment. Movement of glucose into cells is Chassagne P et al: Clinical presentation of hypernatremia in eld accompanied by movement of water out of the intravascular erly patients: A case-control study. This creates a Potassium is the most abundant intracellular cation and the sodium gradient from the tubular lumen into the cell caus second most common cation in the body. The ratio of intra ing passive movement of sodium through epithelial sodium cellular to extracellular potassium concentration is normally channels (enhanced by aldosterone) and generating an elec about 35:1, whereas sodium is much higher in the extracellu tronegative luminal fluid as anions such as chloride and lar space. The importance of these distributions is reflected bicarbonate remain in the lumen. The intracellular:extracel tration and because of the more electronegative fluid. The two major mechanisms that deter (larger quantity of Na+ to draw out of tubule), the presence mine plasma [K+] are renal potassium handling and the dis of poorly reabsorbable tubular anions, and increased intra tribution of potassium between the intracellular and cellular potassium concentration. Failure of these mechanisms for potassium excre tion potentially leads to increased total body potassium in Laboratory determinations of potassium are made on either the face of continued potassium intake. There is no appreciable difference between the two in the absence of thrombocytosis (which can cause Distribution of Total Body Potassium elevated serum potassium but has minimal effect on plasma potassium), and plasma potassium will be used in this dis-the other major mechanism determining potassium balance cussion. Plasma potassium [K+] is closely regulated, but and plasma [K+] is the intracellular-extracellular distribution hyper and hypokalemia do not necessarily indicate of potassium. Only a small quantity of potassium is found in increased and decreased total body potassium because of the the extracellular space. For example, sium is freely distributed throughout the estimated 12 L of movement of K+ out of the cells and into the extracellular extracellular water in a normal 60-kg subject, then only space can mask severe depletion of total body K+; similarly, 48 meq of K+ is present in this space. These pumps are subject to control by insulin and epinephrine, each stimulating increased Na+ and K+ exchange by different mechanisms. Insulin has in fact been Renal Potassium Handling considered by some to have plasma potassium regulation as In the normal steady state, dietary potassium intake is its major role. Clinically, exogenous insulin administration is excreted almost entirely by the kidneys, although small associated with potential for hypokalemia despite no change amounts of potassium are lost in sweat and gastrointestinal in total body potassium. Large amounts of potassium can be excreted by the betics with renal insufficiency (inability to excrete potassium kidneys as long as sufficient time to adapt is given, and potas readily) are prone to severe hyperkalemia unless adequate sium can be conserved with moderate efficiency in normal insulin therapy is given.
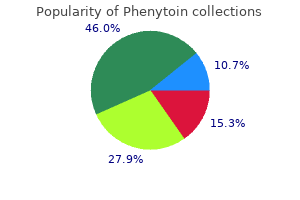
Phenytoin 100mg sale
Another problem with the monoamine hypothesis is the fact that the timing of antidepressant effects on neurotransmitters is far different from the timing of the antidepressant effects on mood medicine reactions order generic phenytoin on-line. The headquarters for the cell bodies of serotonergic neurons is in the brainstem area called the raphe nucleus. Serotonergic projections from raphe to frontal cortex may be important for regulating mood. Serotonergic projections from raphe to basal ganglia may help control movements as well as obsessions and compulsions. Serotonergic projections from raphe to limbic areas may be involved in anxiety and panic. Serotonergic projections to the hypothalamus may regulate appetite and eating behavior. Serotonergic neurons in brainstem sleep centers regulate sleep, especially slow-wave sleep. Serotonergic neurons descending down the spinal cord may be responsible for controlling certain spinal reflexes that are part of the sexual response, such as orgasm and ejaculation. The monoamine receptor hypothesis of depression posits that something is wrong with the receptors for the key monoamine neurotransmitters. Thus, according to this theory, an abnormality in the receptors for monoamine neurotransmitters leads to depression. Depicted here is the normal monoamine neuron with the normal amount of monoamine neurotransmitter and the normal amount of correctly functioning monoamine receptors. Because of these and other difficulties, the focus of hypotheses for the etiology of depression began to shift from the monoamine neurotransmitters themselves to their receptors. As we shall see, contemporary theories have shifted past the receptors to the molecular events that regulate gene expression. Neurotransmitter Receptor Hypothesisthe neurotransmitter receptor theory posits that something is wrong with the receptors for the key monoamine neurotransmitters. According to this theory, an abnormality in the receptors for monoamine neurotransmitters leads to depression. Such a disturbance in neurotransmitter receptors may itself be caused by depletion of monoamine neurotransmitters. The consequences of monoamine neurotransmitter depletion, of stress, or of some inherited abnormality in neurotransmitter receptor could cause the postsynaptic receptors to abnormally up-regulate (indicated in red circle). This up-regulation or other receptor dysfunction is hypothetically linked to the cause of depression. Direct evidence of this is generally lacking, but postmortem studies do consistently show increased numbers of serotonin 2 receptors in the frontal cortex of patients who commit suicide. Indirect studies of neurotransmitter receptor functioning in patients with major depressive disorders suggest abnormalities in various neurotransmitter receptors when using neuroendocrine probes or peripheral tissues such as platelets or lymphocytes. Modern molecular techniques are exploring for abnormalities in gene expression of neurotransmitter receptors and enzymes in families with depression but have not yet been successful in identifying molecular lesions. Depression and Bipolar Disorders 187the Monoamine Hypothesis of Gene Expression So far, there is no clear and convincing evidence that monoamine deficiency accounts for depression; that is, there is no "real" monoamine deficit. Likewise, there is no clear and convincing evidence that excesses or deficiencies of monoamine receptors account for depression; that is, there is no pseudomonoamine deficiency due to the monoamines being there but not the monoamine receptors. On the other hand, there is growing evidence that despite apparently normal levels of monoamines and their receptors, these systems do not respond normally. For instance, probing monoam inergic receptors with drugs that stimulate them can lead to deficient output of neuroendocrine hormones. Such observations have led to the idea that depression may be a pseudomonoamine deficiency due to a deficiency in signal transduction from the monoamine neurotransmitter to its postsynaptic neuron in the presence of normal amounts of neurotransmitter and receptor. If there is a deficiency in the molecular events that cascade from receptor occupancy by neurotransmitter, it could lead to a deficient cellular response and thus be a form of pseudomonoamine deficiency. Such a deficiency in molecular functioning has been described for certain endocrine diseases such as hypoparathy-roidism (parathyroid hormone deficiency), pseudohypoparathyroidism (parathyroid receptors deficient but parathyroid hormone levels normal), and pseudo pseudohy-poparathyroidism (signal transduction deficiency leading to hypoparathyroid clinical state despite normal levels of hormone and receptor). Perhaps a similar situation exists for depression due to a hypothesized problem within the molecular events distal to the receptor. Thus, second messenger systems leading to the formation of intracellular transcription factors that control gene regulation could be the site of deficient functioning of monoamine systems. This is the subject of much current research into the potential molecular basis of affective disorders. This hypothesis suggests some form of molecularly mediated deficiency in monoamines that is distal to the monoamines themselves and their receptors despite apparently normal levels of monoamines and numbers of monoamine receptors. This in turn leads to depression and to the consequences of repeated depressive episodes, namely, more and more episodes and less and less responsiveness to treatment. This possibility that hippocampal neurons are decreased in size and impaired in function during depression is supported by recent clinical imaging studies showing decreased brain volume of related structures. This provides a molecular and cellular hypothesis of depression consistent with a mechanism distal to the neurotransmitter receptor and involving an abnormality in gene expression. Neurokinin Hypothesis of Emotional Dysfunction Another hypothesis for the pathophysiology of depression and other states of emotional dysfunction relates to the actions of a relatively new class of peptide neuro transmitters known as neurokinins (also sometimes called tachykinins). This hypothesis was generated by some rather serendipitous observations that an antagonist to one of the neurokinins, namely substance P, may have antidepressant actions. Classically, substance P was thought to be involved in the pain response because it is released from neurons in peripheral tissues in response to inflammation, causing "neurogenic" inflammation and pain. Furthermore, substance P is present in spinal pain pathways, suggesting a role in central nervous system mediated pain. On the other hand, suggestions that substance P antagonists may have improved mood, if not pain, in migraine patients led to controlled trials of such drugs in patients with depression. This, in turn, leads to depression and to the consequences of repeated depressive episodes, namely, more and more episodes and less and less responsiveness to treatment. This may explain why hippocampal neurons seem to be decreased in size and impaired in function during depression on the basis of recent clinical neuroimaging studies. Classically, substance P was thought to be involved in the pain response because it is released from neurons in peripheral tissues in response to inflammation, causing "neurogenic" inflammation and pain. Substance P and its related neurokinins are present in areas of the brain such as the amygdala that are thought to be critical for regulating emotions. The neurokinins are also present in areas of the brain rich with monoamines, which suggests a potential regulatory role of neurokinins for monoamine neurotransmitters already known to be important in numerous psychiatric disorders and in the mechanisms of action of numerous psychotropic drugs. Thus, antagonists to all three important neurokinins are currently in clinical testing of various states of emotional dysfunction, including depression, anxiety, and schizophrenia. Over the next few years it should become apparent whether this strategy can be exploited to generate truly novel psychotropic drugs acting on an entirely new neurotransmitter system, namely, the neurokinins. Substance P and its related neurokinins are present in areas of the brain such as the amygdala that are thought to be critical for regulating emotions. The neurokinins are also present in areas of the brain rich in monoamines, which suggests a potential regulatory role of monoamine neurotransmitters, which are already known to be important in numerous psychiatric disorders and in the mechanisms of action of numerous psychotropic drugs. The first neurokinin was discovered in the 1930s in extracts of brain or intestine. This molecule is now known to be a string of 11 amino acids (an undecapeptide). This is in sharp contrast to monoamine neurotransmitters, which are modifications of a single amino acid. The following are some of the differences between the synthesis of neurotrans mitter by a monoaminergic neuron and by a peptidergic neuron. Whereas mono amines are made from dietary amino acids, peptide neurotransmitters are made from proteins that are direct gene products. However, genes are not translated directly into peptide neurotransmitters but into precursors of the peptide neurotransmitters. Further modifications convert these grandparent proteins into the direct precursors of peptide neurotransmitters, sometimes called the "parents" of the neuropeptide, or the propeptides. Finally, modifications of the parental peptide produces the neuropeptide progeny itself. Finally, this protein is clipped even shorter by another enzyme, called a converting enzyme, in the synaptic vesicle and forms substance P itself.

100 mg phenytoin fast delivery
Respiratory failure is usually of the hypercapnic variety unless there is atelectasis or consolidation from pneumonia medications bipolar discount 100 mg phenytoin fast delivery. Thoracic Wall Disorders Mechanical ventilation is often necessary to perform the work of breathing in the patient with muscle weakness who develops hypercapnia. Lung compliance and resistance are normal in the absence of secondary pulmonary complications. As in patients with obstructive or interstitial lung dis Paco >50 mm Hg, usually with hypoxemia. If respiratory muscle weakness is the primary problem but ventilatory control is intact, pressure General Considerations support ventilation may be suitable. However, acquired and congenital abnormalities may the inspiratory muscles continue to contract throughout result in distortion of the chest wall, mechanical disadvan inspiration, whereas if the ventilator initiates the breath, tage of the respiratory muscles, increased work of breathing, inspiration is completely passive on the patients part. Patients usually have Pressure-support ventilation, although the inspiratory mus chronic respiratory failure with hypoxemia, hypercapnia, cles are not completely rested, may have potential advantages and cor pulmonale but also may present with acute deterio of allowing some work to be done but with less effort. The most common chronic disor In some patients, positive-pressure ventilation is needed ders leading to respiratory failure are severe obesity of the only for some part of the day eg, (at night), and the patient can chest wall and abdomen, kyphoscoliosis and scoliosis, and breathe well for long periods of time. Massive ascites, late-stage pregnancy, necessary to attach the ventilator at night, or noninvasive ven recent abdominal or thoracic surgery and trauma, and tilation may be tried for intermittent use. Bednarik J, Lukas Z, Vondracek P: Critical illness polyneuromy opathy: the electrophysiological components of a complex Pathophysiology entity. Obese patients are more prone to develop obstructive Respiratory failure results from a combination of increased sleep apnea because of a greater redundant soft tissue in the work of breathing and ventilation-perfusion maldistribution upper airway, potentiating airway obstruction during sleep owing to restriction of expansion of the lungs and chest wall. A syndromethe mechanics of the chest wall and the diaphragm are of obesity-hypoventilation has been described. The mecha altered in ways unique to the type of disorder and the loca nism of this central hypoventilation is unclear but is proba tion of the abnormalities. These patients have daytime hypercapnia from idiopathic or due to poliomyelitis, tuberculosis, or other iden depressed chemoresponsiveness, increased work of breath tifiable causes. Patients with scoliosis have an inverse relation ing, and abnormal pulmonary function. Patients with ship between the severity of scoliotic deformity and the obesity-hypertension syndrome often have pulmonary compliance of the thoracic cage. Most often they have basilar atelecta Ankylosing spondylitis limits rib cage expansion, and sis, low tidal volumes, and mild hypoxemia. If diaphragmatic patients tend to breathe primarily by diaphragmatic move excursion is severely limited, these patients may develop ment as the thoracic cage becomes increasingly immobile. The underlying condition may contribute tothe contribution of thoracic cage expansion to tidal breath additional problems. For example, patients with liver disease, ing falls as minute ventilation increases. The position of the ascites, or tumors also may have pleural effusions, further thoracic cage at end expiration often becomes fixed at a vol compromising lung function. The chest wall therefore trauma may be unable or unwilling to take deep inspirations exerts a greater than normal outward pull on the lungs, result because of pain. The flail is the paradoxical inward movement of the ventilation-perfusion maldistribution contributing to chest wall during inspiration, making ventilation inefficient hypoxemia and hypercapnia. Chest pain and diaphragmatic excursion, this finding has been attributed underlying lung contusions contribute to respiratory failure. Patients 2 burden on the chest wall and the diaphragm during inspira with ascites and abdominal tumors would be expected to be tion. In scoliosis and spondylitis, regional differences in the chest wall and diaphragm to be more inward than nor ventilation can be explained by differences in expansion mal. Thus the amount of gas that can be further expired from owing to local chest wall stiffness in spondylitis or asymmet the end-expiratory position (expiratory reserve volume ric deformity in scoliosis. The work of breathing is increased, especially when For reasons that are not clear, cor pulmonale is seen more minute ventilation increases. The larger mass of the chest often in chronic respiratory failure from scoliosis, sometimes wall and abdominal wall must be accelerated at each breath, in the absence of severe gas-exchange abnormalities; it may and additional energy is expended to move them during tidal be related to anatomic deformity or arrested development of breathing. It is likely that Clinical Features obesity associated with more peripheral distribution of added adipose tissue (eg, buttocks and extremities) has less Some patients with thoracic wall abnormalities have chronic effect on respiratory function, but this has not been studied. The due to further abrupt worsening of the chest wall disease, chronicity of respiratory failure can be confirmed by finding development of a lung complication such as pneumonia or an elevated plasma bicarbonate level. Patients present usually with rapid, liosis and increases with increasing scoliosis. As many as 50% shallow tidal breathing unless there is a disorder of ventila of patients with an angle greater than 80 degrees may be con tory control suggesting central hypoventilation. An angle of 100 degrees examination can show obvious chest wall deformity or seems to be associated with dyspnea on exertion and an angle decreased range of motion of the chest wall or diaphragm, greater than 120 degrees with alveolar hypoventilation. Wheezing, rhonchi, or stridor may Treatment be evidence of superimposed obstructive airway disease such as asthma, but kinking of large central and upper airways in Care of the patient with respiratory failure from thoracic wall scoliosis may be the cause. There is considerable tions such as pneumonia, left-sided heart failure, atelectasis, reported success with noninvasive positive-pressure ventila and pleural effusions. Lung persist, likely because of improvements in respiratory muscle volumes do not decrease routinely in proportion to excess function and improved respiratory gas exchange. Mechanical weight, and no marker of obesity such as weight/height2 ventilation is initiated if the patient requires additional ven (body mass index) predicts respiratory failure. For example, tilatory support to overcome increased metabolic require in otherwise normal subjects who are more than 160% of ments or during an acute exacerbation owing to infection or ideal weight, lung volumes and flows are usually found to be surgery. Such normal lung function may not persist, how to respiratory failure, patients often will respond after several ever, as these patients age, and the decline in lung function days of correction of hypoxemia and respiratory acidosis by with advancing age may be accelerated compared with demonstrating greatly improved respiratory drive. In addition, obesity is associated with Respiratory stimulant drugs such as progesterone and hypertension and cardiovascular disease, which themselves almitrine are not helpful. Central matic excursion are approached according to the type of hypoventilation in obesity (obesity-hypoventilation syn problem. Those with severe ascites may benefit from large drome) further dissociates the degree of obesity from the volume paracentesis or other efforts to decrease the volume severity of hypercapnia, as does the high prevalence of basi of ascites. Pain management is critical in patients following lar atelectasis causing hypoxemia. Patients with ascites from liver disease and pregnancy fre Treatment of flail chest depends on the severity of injury. The severity of respiratory impairment ratory failure develops, mechanical ventilation may be neces from decreased diaphragmatic excursion is highly unpre sary until chest wall mechanics recover. Only in rare cases is dictable, but most of the patients have some degree of hypox surgical repair indicated, including prolonged recovery, emia and only rarely develop hypercapnia. As the disorder progresses, sitive to preload reduction and rapid decrease of intravascu work of breathing increases. Other patients will respond with improved gas those without chronic respiratory failure. Although precipi exchange after a short period of supplemental oxygen or tated most often by exacerbation of airway obstruction from mechanical ventilation. While there is little chance of improv infection and increased sputum production, an important ing the underlying pathophysiology in severe scoliosis or factor leading to acute respiratory failure is inspiratory mus severe ankylosing spondylitis, weight reduction and treatment cle fatigue. During expiration, decreased lung elastic recoil lowers the distending pressure holding the Chronic Obstructive Pulmonary Disease airways open and leads to increased airway resistance. The mechanism of lung Increased dyspnea and cough; decreased exercise parenchymal destruction in the majority is unknown. Many patients volume, increased sputum purulence, and/or worsen with emphysema have clinical features of chronic bronchitis.
Order phenytoin visa
Net phosphate excretion is prima movement into cells (facilitated by insulin) and subsequent rily through the kidneys by filtration and reabsorption symptoms 8 weeks purchase phenytoin 100mg with visa. The most striking examples of rapid, mal tubules determines phosphorus excretion, and this severe falls in plasma phosphorus are seen in the treatment mechanism is driven by proximal tubular sodium reabsorp of diabetic ketoacidosis and in the refeeding syndrome. Thus there is enhanced phosphorus reabsorption in Diabetic ketoacidosis is associated with pretreatment extra the face of increased proximal sodium reabsorption in cellular phosphate loss from solute diuresis. However, proximal phosphorus tion of insulin results predictably in hypophosphatemia as reabsorption is also independently regulated by the parathy glucose and phosphate move into cells. This can lead to dissociation between plasma phosphate during enteral or parenteral refeeding of sodium reabsorption and phosphorus reabsorption, as in chronically malnourished individuals, including alcoholics, hyperparathyroidism. Respiratory alkalosis also causes a shift of extracellular Hypophosphatemia phosphorus into cells. In and rhabdomyolysis may occur with plasma phosphorus addition, binding of phosphorus in the gastrointestinal tract <1 mg/dL. In critically ill Fat malabsorption patients, renal phosphate excretion increases with solute Chronic antacid use diuresis and with the use of acetazolamide, a carbonic anhy Acute redistribution of extracellular phosphorus drase inhibitor. Metabolic acidosis increases the release of Respiratory alkalosis Sepsis inorganic phosphate into the extracellular space, resulting in Salicylate toxicity increased renal excretion of phosphate, but this is not usually Hepatic encephalopathy a cause of hypophosphatemia because phosphorus can be Toxic shock syndrome mobilized easily from the intracellular stores. Hemodialysis Glucose-insulin administration is a relatively inefficient way of removing phosphate; there Diabetic ketoacidosis fore, hypophosphatemia is an unusual complication of renal Refeeding syndrome replacement therapy. Erythrocyte inorganic phosphate have a combination of respiratory alkalosis, pain, sepsis, and concentration is directly related to plasma phosphorus, and increased tissue uptake of phosphate. Patients with severe inorganic phosphate is required for the conversion of glycer head injury are reported to have hypophosphatemia and aldehyde 3-phosphate to 1,3-diphosphoglyceric acid, a key hypomagnesemia owing to excessive urinary losses. In hypophosphatemia, glycolytic interme diates preceding this enzymatic step accumulate and those A. Patients may have difficulty wean throcyte glycolytic enzyme deficiencies such as pyruvate ing from mechanical ventilation or may present with symp kinase deficiency. Impaired function of skeletal muscles, toms and signs of congestive heart failure. In one study, decreased respi result in an increased tendency to infection, and platelet dys ratory and peripheral muscle phosphate concentrations were function may contribute to bleeding. Findings have included changes in mental status, seizures, Clinical Features and neuropathy. Although most patients with hypophosphatemia are identi fied by routine monitoring of electrolytes, hypophosphatemia B. Other laboratory findings may include features dates for symptomatic hypophosphatemia are those with of hemolysis, elevated creatine kinase, and qualitative combinations of mechanisms, such as patients with diabetic platelet dysfunction (prolonged bleeding time) when ketoacidosis with solute diuresis who are receiving insulin plasma phosphorus is 0. In is most useful; arterial blood gases and plasma glucose, patients receiving intravenous glucose, phosphorus supple electrolytes, and calcium may be helpful. Routine repletion of phosphorus in patients with diabetic Treatment ketoacidosis has been recommended because of the high fre A. It has been proposed that hypophos require immediate treatment if weakness involving the phatemia contributes to decreased oxygen delivery, insulin respiratory muscles precipitates respiratory failure. This is espe diate or final outcome from routine phosphate replacement cially important when a further decrease in phosphorus is has not been demonstrated. Supportive care is essential while severe hypophos sive phosphate repletion include volume overload from phatemia is corrected. In older patients with elemental phosphorus and phosphate concentrations and renal insufficiency and small children, especially with fluid amounts are expressed. Therefore, close monitoring of plasma phosphorus Ritz E, Haxsen V, Zeier M: Disorders of phosphate metabolism: and other electrolytes is necessary during repletion, espe Pathomechanisms and management of hypophosphataemic dis cially if phosphate is given as the potassium salt. For a 60-kg adult, this would be approximately 400 mg phosphorus, or Hyperphosphatemia about 4 mL of sodium or potassium phosphate solution (3 mmol/mL) in the 1-L infusion. In less severe hypophosphatemia, an appropriate starting Usually no acute symptoms. Oral Cardiac conduction system disturbances and features of supplementation can be provided using potassium phosphate hypocalcemia may occur. Factors that may lead to this situation include renal insufficiency and continued replace Hyperphosphatemia as a clinical problem is most often the ment of phosphorus after reversal of the cause of hypophos result of long-standing elevation of plasma phosphorus con phatemia. Enemas or oral bowel phate salts in the heart, kidneys, and lungs; rarely, acute car preparation products used prior to radiographic procedures diac conduction disturbances can occur. In addition, calcium or colonoscopy may contain a large quantity of sodium phosphate precipitation results in acute hypocalcemia and its phosphate as an osmotic agent. Rarely, in patients given large amounts of sodium phosphate as a cathartic or enema, severe anion gap metabolic acidosis may result. Clinical Features Patients in whom this has been reported are elderly or very Patients at high risk for development of hyperphosphatemia young and often have renal insufficiency. In that is taken back up into the cells or bone or excreted by more severe cases, if the calcium phosphorus product is the kidney. Impaired excretion primarily results from greater than 60, the risk of ectopic calcification in various chronic renal insufficiency, and because parathyroid hor organs increases, including the heart, lungs, and kidneys. Plasma calcium should be monitored dur sue breakdown, a form of redistribution of a large ing treatment of both hypo and hyperphosphatemia. Tumor lysis syndrome is seen Chronic renal failure uncommonly in patients with solid tumors, except those Acute renal failure Extracellular volume depletion with extensive necrosis. Bowel necrosis from ischemia also Hypoparathyroidism may be associated with hyperphosphatemia. Renal insuffi Acute redistribution of intracellular phosphorus ciency exacerbates hyperphosphatemia caused by redistri Massive tissue breakdown bution of phosphorus. Because insulin and glucose drive Rhabdomyolysis phosphorus into cells, diabetics with insulin deficiency Tumor lysis syndrome (lymphoma) also may be more prone to hyperphosphatemia, but this is Exogenous phosphorus intake rarely significant. Excessive treatment of hypophosphatemia Increased dietary phosphorus (with renal insufficiency) C. In rhabdomyolysis, plasma creatine kinase and aldolase are elevated, and myoglobinuria may be present. The distribution of magnesium is similar to that of Treatment potassium, with the vast majority (99%) of magnesium resid ing inside cells. Magnesium plays elevated plasma concentration of phosphorus that requires an important role in neuromuscular coupling, largely immediate treatment. Among the causes of hypomagnesemia are drugs fre hyperphosphatemia should be treated by lowering the quently used in critically ill patients such as amphotericin B, plasma phosphorus concentration rather than by adminis diuretics, and aminoglycoside antibiotics, but hypomagne tration of calcium because the latter action may worsen semia is also seen in malnutrition, chronic alcoholism, and ectopic calcification.
Diseases
- Cockayne syndrome type 3
- Aplasia cutis congenita of limbs recessive
- Nephrosis neuronal dysmigration syndrome
- Laryngeal cleft
- Scott syndrome
- Thalassemia major
- Bronchiectasis
Purchase phenytoin 100 mg mastercard
Later medications kosher for passover purchase cheap phenytoin on line, once the reader is familiar with the general concepts outlined here, these scientific strategies will be applied in the subsequent chapters to many specific psychiatric and neurological disorders. We will then show how these three approaches can be applied to learning how modifications in chemical neurotransmission might lead to various brain disorders. Specific concepts that will be explained are the molecular neuro biology and genetics of psychiatric disorders, neuronal plasticity, and excitotoxicity. Obviously, one must first understand normal brain functioning and normal chemical neurotransmission in order to have any chance of detecting, let alone understanding, neurobiological abnormalities that cause psychiatric and neurological disorders. Neurobiology Limited definitionthe study of brain and neuronal functioning Approach Studies using experimental animals Use of drugs to probe neurobiological and molecular regulatory mechanisms Findings relevant to psychopharmacology Discovery of neurotransmitters and their enzymes and receptors Principles of neurotransmission Genetic and molecular regulation of neuronal functioning Neurobiological regulation of animal behaviors trols this whole process. Many of the lessons derived from this approach have already been discussed in the preceding chapters. However, the importance of pursuing the causes of mental disorders is underscored by how frequent these illnesses are in our society and how limited current treatments for them can be. That is, as many as one in five persons may experience a mental illness during their lifetimes, and about 4% of the population has a chronic and severe mental disorder. Furthermore, currently available treatments in psychopharmacology are not strictly "curative" but are merely palliative, reducing symptoms without necessarily offering sustained relief. Better treatments of the future now depend on discovering the causes of mental illness. This discipline uses the results of neurobiological investigations of normal brain functioning as a basis for the search for the substrate of abnormal brain functioning in psychiatric disorders. Scientists have long suspected that abnormalities in brain enzymes or receptors are major contributors to the causes of mental illness and have been searching for an enzyme or receptor deficiency that could be identified as the cause of specific psychiatric disorders. Such tools available for use in humans include studies of enzymes, receptors, and genes in postmortem brain tissues and in peripheral tissues that can be ethically 102 Essential Psychopharmacology Table 4-2. Metabolites of neurotransmitters can be studied in cerebrospinal fluid, plasma, and urine. Receptors for neurotransmitters can also be studied indirectly by using selective drug probes, which cause hormones to be released into the blood that can be measured and therefore serve as a reflection of brain receptor stimulation. Unfortunately, little progress has been made yet in defining the biological causes of mental illnesses by using these approaches. No single reproducible abnormality in any neurotransmitter or in any of its enzymes or receptors has been shown to cause any common psychiatric disorder. Indeed, it is no longer considered likely that one will be found, given the complexity of psychiatric diagnosis and the profound interaction of environmental factors with genetics in psychiatric disorders. More Chemical Neurotransmission as the Mediator of Disease Actions 103 recently, biological psychiatry has shifted from a strategy of pursuing a single unique biochemical lesion as the cause of each psychiatric disorder to the discovery and enumeration of risk factors that do not cause illness by themselves but contribute to the risk of a psychiatric disorder. This approach is sometimes called complex genetics because it is indeed complicated, as we shall see below. The potential usefulness of this approach is underscored by findings from genetic studies of mental illnesses. Despite strong evidence from twin studies that genetic susceptibility exists for both bipolar disorder and schizophrenia, no specific gene has been unambiguously identified for the usual forms of any common mental disorder. Rather, the genetics of major psychiatric disorders are likely to be at best contributors in multiple complex ways to these illnesses, just as is currently suspected for coronary artery disease, diabetes, and hypertension. Methods to approach the complex genetics of mental illnesses are just evolving and include such techniques as linkage, linkage disequilibrium, and association studies to name a few. Rather, a whole list of abnormally acting genes and their corresponding gene products, triggered by both inherited and acquired risk factors, are hypothesized to act together or in just the right sequence to cause clusters of symptoms that appear in different psychiatric disorders. Once the complete list of genes and environmental factors that comprise all the vulnerabilities to a psychiatric illness is determined, it will be necessary to understanding how all the corresponding gene products participate in the neuronal functioning and especially the chemical neurotransmission that mediate the mental illness. The long-term hope, of course, is that by knowing this, a logical biochemical rationale can be found for reversing these abnormalities with drug therapies. The question of how this could lead to a rational drug therapy to halt, reverse, or compensate for these multiple simultaneous biochemical events leaves us in a complete quandary at present. It might be possible to pursue treatments based on this knowledge if the abnormal gene products proved to be enzymes or receptors that could be stimulated or blocked by drugs. However, it is not likely to be this simple, as multiple simultaneous drugs acting to compensate for each genetic abnormality that contributes to the disease vulnerability might prove to be necessary. At any rate, the biological psychiatry hunt is on, but treatments based on this approach certainly do not appear to be right around the corner. Psychopharmacology Limited definitionthe use of drugs to treat symptoms of mental illnessthe science of drug discovery, targeting enzymes and receptors Approach Studies in patients with psychiatric disorders Serendipitous clinical observations In clinical investigations, the use of drugs with known mechanisms of action to provoke biological or behavioral responses that would provide clues where abnormalities in brain functioning may exist in specific psychiatric disorders In drug discovery, theory-driven targeting of enzymes and receptors hypothesized to regulate symptoms in a psychiatric disorder Psychopbarmacological results In clinical investigations, the first observation is often a serendipitous discovery of clinical efficacy, after which the biochemical mechanism of action is discovered In drug discovery, specific enzymes or receptors are first targeted for drug action. That is, if a drug with a well understood mechanism of action on a receptor or enzyme causes reproducible effects on the symptoms of a patient with a brain disorder, it is likely that those symptoms are also linked to the same receptor that the drug is targeting. Using drugs as tools in this manner can help map which receptors and enzymes are linked to which psychiatric or neurological disorder. Since drug actions are much better known than disease actions at the present time, the use of drug tools in this manner has so far proved to be the more productive approach to understanding diseases as compared with the biological psychiatry approach of looking for abnormal receptors, enzymes, or genes. Indeed, much of what is known, hypothesized, or theorized about the neurochemical abnormalities of brain disorders is derived from the approach of using drugs as tools. Thus, pathophysiology is inferred rather than proved, since we do not yet know the primary enzyme, receptor, or genetic deficiency in any given psychiatric or neurological disorder. These theories, in fact, direct the biological psychiatry researcher where to look for proof of disease abnormalities. Thus, psychopharmacology is bidirectional in the sense that certain drugs, namely, those that have a known neurochemical mechanism of action and that are also effective in treating brain disorders, help to generate hypotheses about the causes of those brain disorders. The other direction of psychopharmacology is that in the case of a brain disorder with a known or suspected pathophysiology, drugs can be rationally designed to act on a specific receptor or enzyme to correct the known or suspected pathophysiology and thereby treat the disorder. It would be advantageous for new drug development to proceed from knowledge of pathophysiology to the invention of new therapeutics, but this must await the elucidation of such pathophysiologies, which, as emphasized here, are yet largely unknown. Virtually all effective psychopharmacological drugs that have been discovered to date were found by serendipity (good luck) or by empiricism, that is, by probing disease mechanisms with a drug of known action but no prior proof that such actions would necessarily be therapeutic. Hopefully, a rational route from pathophysiology to drug development will become increasingly available as the molecular causes of such disorders are elucidated in coming years. A new approach to selecting specific drugs for individual patients called phar macogenetics is dawning in psychopharmacology. Although in its infancy, pharmaco genetics attempts to match the likelihood of a positive or negative clinical response to a given drug with the specific genetic makeup of the patient. The idea is that knowing the critical genetic information about a patient, not just the psychiatric diagnosis, could lead to a more rational decision as to which drug to prescribe for that patient. Currently, there is no rational way to predict which antidepressant is more likely than another to work in any depressed patient or which antipsychotic would be best for a given schizophrenic patient. Perhaps certain genetic characteristics will predict the likelihood of a better therapeutic response or better tolerability of one drug over another. To date, no such genetic factors are yet known that can assist the prescriber in selecting psychotropic drugs for individual patients. How Synaptic Neurotransmission Mediates Emotional Disorders Despite a frustrating lack of knowledge of specific pathophysiological mechanisms for various psychiatric disorders, a good deal of progress has been made in our thinking about mechanisms whereby synaptic neurotransmission can mediate disease processes. Discussed below are several general concepts relating to how psychiatric disorders are thought to be associated with modifications in synaptic neurotransmission. Geneticists no longer talk about inheriting a mental illness; they talk about inheriting vulnerability to a mental illness. Such vulnerability theoretically arises from a set of abnormally functioning genes, and some of this abnormal functioning is inherited. Since genes control all functions of the neuron, all psychiatric disorders at some level are genetic. However, that does not necessarily mean that all abnormal functions of genes are inherited. Vulnerability factors for psychiatric disorders are as yet poorly understood, multiple in number, and very complicated. Nevertheless, a few important principles of genetic vulnerability have been established.

Purchase phenytoin 100 mg amex
G Gastrointestinal: Hypercalcemia associated symptoms include anorexia symptoms mold exposure purchase phenytoin on line amex, nausea, vomiting, constipation, and peptic ulcer disease. G Psychiatric and neurocognitive: Patients may have depressed mode, lethargy, emotional lability, and decreased cognitive function. Imaging G Sestamibi scan: 99mTc sestamibi localizes to the mitochondria of para thyroid cells, which are rich in mitochondria. Disadvantages include difficulty of localization of nonstandard locations and the potential of confusion with thyroid abnormalities, and interoperator variability. G Selective venous sampling:the veins draining the parathyroid region can be sampled. Low serum phosphorus, increased 24-hour urinary calcium excretion, elevated serum 1,25-dihydroxyvitamin D may be seen. It is important to rule out familial hypocalciuric hypercalcemia because usually the course of this disease is benign and parathyroidectomy is not indicated. Past medical history should be carefully obtained as these patients are asymptomatic and have a history of elevated calcium levels since childhood. Secondary hyperparathyroidism should also be ruled out (either from a renal source or from decreased calcium absorption/intake or vitamin D deficiency). Treatment Options Medical Medical treatment is indicated in patients who do not meet the criteria for surgery, refuse surgery, or are poor surgical candidates. Medications used in the treatment of osteoporosis, such as bisphosphonates, may be useful. Surgical Surgery is curative and is indicated in all cases with symptomatic dis ease. Following are the indications of surgery in asymptomatic patients: (1) serum calcium "1. Preoperative imaging localization allows for guided and minimally invasive parathyroi dectomy in most cases. Common ectopic sites include the thymus/mediastinum, transesophageal groove, retroesophageal, intrathyroidal, and the carotid sheath. Head and Neck 499 this disease is more likely to be associated with profound hypercalcemia or hypercalcemic crisis. In a bilateral neck exploration, identify all four glands; perform a subtotal or total parathyroidectomy with thymectomy and autotransplantation of gland as needed. It is controversial if a subtotal or total parathyroi dectomy should be performed. After a bilateral exploration, a subtotal or total parathyroidectomy with auto transplantation can be done. Treatment consists of treating the initial cause of the secondary hyperparathyroidism. N Tertiary Hyperparathyroidism Tertiary hyperparathyroidism is due to prolonged hypercalcemia that causes parathyroid gland hyperplasia. Most commonly seen after renal transplant for end-stage renal disease that is associated with severe secondary hyperparathyroidism. Stuttgart/ New York: Thieme; 2009:197225 500 Handbook of OtolaryngologyHead and Neck Surgery 5. N Etiology G Iatrogenic (surgical): this is the most common cause of hypoparathyroid ism. This may occur after surgery on the neck (thyroidectomy, parathy roidectomy, or neck dissection). It usually occurs as a result of manipulation of blood supply to the parathyroid glands during surgery or injury to or removal of one or more parathyroid glands. G Hypoparathyroidism may also occur in the setting of hungry bone syndrome,following surgery. This is usually associated with severe preoperative hy perparathyroid bone disease. G Autoimmunity: this occurs secondary to immune-mediated destruction of parathyroid glands. G Hypoparathyroidism may occur due to abnormal development of the parathyroid glands. This is usually associated with DiGeorge syndrome, which is a congenital abnormality of the third and fourth branchial pouches and results in the absence of parathyroid glands and thymus. G Hypoparathyroidism may result from the destruction of parathyroid glands due to irradiation or infiltrative diseases (such as metastatic cancer and granulomatous disease) or storage diseases (such as Wilsons disease and hemochromatosis). N Clinical Signs and Symptoms Patients present with symptoms and signs of hypocalcemia (see Chapter 5. Differential Diagnosis Hypoparathyroidism is usually associated with hypocalcemia (low serum calcium levels), hyperphosphatemia (high phosphate levels), and low or 5. Note that, in contrast, hungry bone syndromeis associated with hypocalcemia, and hypophosphatemia (low phosphate levels). Patients with hypoparathyroidism may require lifelong supplementation with calcium and vitamin D, except for those with transient hypoparathyroidism. The goal of treatment is to maintain serum calcium levels in the low normal range. G Hypocalcemia occurs in patients with renal failure, vitamin D deficiency, magnesium deficiency, acute pancreatitis, and with hypoparathyroidism and pseudohypoparathyroidism. A basic 502 Handbook of OtolaryngologyHead and Neck Surgery metabolic panel and a comprehensive metabolic panel measure the total calcium levels (bound plus unbound), although it is the free (unbound) form that is most important. N Control of Calcium Metabolism Calcium is absorbed from the gut, stored in the bone, and excreted by the kidneys. G Calcitonin: Calcitonin is synthesized in the C cells of the thyroid and causes a decrease in plasma calcium and phosphate levels. N Hypocalcemia Etiology G Hypoparathyroidism:the most common cause is iatrogenic (surgery); see Chapter 5. G Renal failure G Vitamin D deficiency G Hypomagnesemia G Acute pancreatitis G Hyperphosphatemia Clinical G Neuromuscular irritability: Patients may present with tingling, par esthesias in fingers and periorally, tetany, carpopedal spasm, seizures, irritability, and confusion. G Other manifestations include subcapsular cataracts, dry flaky skin, and brittle nails. Head and Neck 503 Evaluation Serum albumin concentration should be measured in patients with hypocalcemia. When albumin is low, the calcium level should be corrected (adjusted) for the level of albumin. G Acute hypocalcemia: Patients with symptomatic tetany, seizures, or stridor need immediate correction of hypocalcemia. G Chronic hypocalcemia: Calcium and vitamin D supplementation are the mainstays of treatment. Vitamin D supplementation is given as ergocalciferol (vitamin D2), which has a long duration of action, or calcitriol (Rocaltrol), which has a short duration of action. N Hypercalcemia Etiology G Hyperparathyroidism is the most common cause of hypercalcemia; see Chapter 5. Other lab values should be obtained to diagnose the suspected underlying etiology. Treatment Options Treatment should focus on the underlying etiology of hypercalcemia. Use with caution in patients with heart failure or renal failure to avoid fluid overload. The serum electrolytes, calcium, and magnesium levels should be monitored closely. Once the fluid deficit has been corrected, consider adding furosemide (loop diuretic). Bisphosphonates can be given as these inhibit bone resorption by osteoclasts and are effective in lowering calcium levels to the normal range within 2 to 5 days. Calcitonin rapidly lowers serum calcium and can be administered subcutaneously as an adjunct. G Chronic severe hypercalcemia:the cause should be identified and treated accordingly (see Chapter 5. Bisphosphonates, gallium nitrate, or glucocorticoids can be used in the treatment of malignancy associated hypercalcemia. Hypercalcemia associated with granulomatous diseases is also treated with glucocorticoids. If hypercalcemia is caused by medi cation overdose, then that medication should be stopped.

Best purchase for phenytoin
There is less weight gain with risperidone than with some other atypical antipsychotic agents medications grapefruit interacts with cheap phenytoin 100 mg otc, perhaps because risperidone does not block histamine 1 receptors, but weight gain is still a problem for some patients. Olanzapine Although olanzapine has a chemical structure related to that of clozapine. Olanzapine lacks the extreme sedating properties of clozapine but can be somewhat sedating. Olanzapine is associated with weight gain, perhaps because of its antihistaminic and serotonin 2C antagonist properties. Early studies suggest a very low incidence of tardive dys kinesia with long-term use and also suggest that some patients improve with olanzapine when conventional antipsychotics fail, although probably not as much as they would with clozapine. Many studies demonstrate that olanzapine is highly effective for positive symptoms of schizophrenia and also improves negative symptoms of schizophrenia better than do conventional antipsychotics. Ongoing studies also show that olanzapine improves mood, not only in schizophrenia but also in the manic and depressed phases of bipolar disorder, suggesting that it may be a first-line treatment for bipolar disorder, as discussed in Chapter 7. Some studies suggest that olanzapine may improve cognitive functioning in schizophrenia and in dementia. Quetiapine Quetiapine also has a chemical structure related to that of clozapine. It is also useful in schizophrenia, bipolar disorder, and other types of psychosis, in which it has few extrapyramidal side effects. It has shown species-specific inhibition of cholesterol biosynthesis in the lens of some animals, where it can cause cataracts, but is not documented to do this in humans. Some patients improve on quetiapine when conventional antipsychotics fail, although probably not as well as on clozapine. Studies demonstrate that quetiapine is highly effective for the positive symptoms and also improves the negative symptoms of schizophrenia. Ongoing studies are beginning to show that quetiapine may improve mood in schizophrenia and in the manic and depressed phases of bipolar disorder. Some studies suggest that quetiapine may improve cognitive functioning in schizo phrenia and also in dementia. Also, ziprasidone is the only atypical antipsychotic that is a serotonin 1D antagonist, a serotonin 1A agonist, and also inhibits both serotonin and norepinephrine reuptake. Some patients improve with ziprasidone when conventional antipsychotics fail, although probably not as much as with clozapine. Studies demonstrate that ziprasidone is highly effective for the positive symptoms and also improves the negative symptoms of schizophrenia. Some studies suggest that ziprasidone may improve cognitive functioning in schizophrenia and also in dementia. Pharmacokinetic Considerations for the Atypical Antipsychotic Drugs Many of the general principles of pharmacokinetics were introduced in Chapter 6 on antidepressants (see also. Here we will discuss some specific pharmacokinetic issues relating to antipsychotic drugs. It has a unique pharmacological profile, different from those of all other atypical antipsychotics. As with all atypical antipsychotics discussed in this chapter, binding properties vary greatly with technique and species and from one laboratory to another; they are constantly being revised and updated. Two atypical antipsychotic drugs are substrates of 1A2, namely olanzapine and clozapine. As with all atypical antipsy chotics discussed in this chapter, binding properties vary greatly with technique and species and from one laboratory to another; they are constantly being revised and updated. Although this may not be particularly clinically important for olanzapine (other than causing slightly increased sedation), it could potentially raise plasma levels sufficiently in the case of clozapine to increase the risk of seizures. Thus, the dose of clozapine may need to be lowered when administering it with fluvoxamine, or another antidepressant may need to be chosen. On the other hand, when an inducer of 1A2 is given concomitantly with either of the two antipsychotic substrates of 1A2, the level of the antipsychotic may fall. This happens when a patient begins to smoke, because smoking induces 1A2, and this would cause levels of olanzapine and clozapine to fall. Theoretically this might cause patients stabilized on an antipsychotic dose to relapse if the levels fell too low. Also, cigarette smokers may require higher doses of these atypical antipsychotics than nonsmokers. Cytochrome P450 2D6 Another cytochrome P450 enzyme of importance to atypical antipsychotic drugs is 2D6. When these drugs are given with an inhibitor of this enzyme, such as the antidepressant fluvoxamine, plasma levels of olanzapine and clozapine can rise. For risperidone the clinical significance of this is uncertain, since both the parent drug and the metabolite are active. Theoretically, the dose of olanzapine and clozapine may have to be lowered when given with an antidepressant that blocks 2D6, although this is not often necessary in practice. Several psychotropic drugs are weak inhibitors of this enzyme, including the antidepressants fluvoxamine, nefazodone, and norfluoxetine, which is an active metabolite of fluoxetine. For the four atypical antipsychotics that are metabolized by 3A4, the clinical implication is that concomitant administration with a 3A4 inhibitor may require dosage reduction of the atypical antipsychotic. Drugs can not only be substrates for a cytochrome P450 enzyme or an inhibitor of a P450 enzyme, they can also be inducers of a cytochrome P450 enzyme and thereby increase the activity of that enzyme. Since mood stabilizers may be frequently mixed with atypical antipsychotics, it is possible that carbamazepine may be added to the regimen of a patient previously stabilized on clozapine, quetiapine, ziprasidone, or sertindole. If so, the doses of these atypical antipsychotics may need to be increased over time to compensate for the induction of 3A4 by carbamazepine. On the other hand, if carbamazepine is stopped in a patient receiving one of these four atypical antipsychotics, the antipsychotic dose may need to be reduced, because the autoinduction of 3A4 by carbamazepine will reverse over time. Atypical Antipsychotics in Clinical Practicethe atypical antipsychotics are still relatively new, particularly some members. Information about new drugs is first available from clinical trials and then modified by observations from clinical practice, and the atypical antipsychotics are no exception to this pattern. Some findings from clinical practice have already confirmed those from clinical trials for the three marketed atypical antipsychotics. Second, atypical antipsychotics reduce negative symptoms of schizophrenia better than do conventional antipsychotics, but this may be because they do not make things worse as much as because they really reduce negative symptoms. Third, atypical antipsychotics reduce affective symptoms in schizophrenia and related disorders such as treatment-resistant depression and in bipolar disorder, where treatment effects appear to be quite robust. The magnitude of these properties is far from trivial and, in fact, makes the four atypical antipsychotics risperidone, olanzapine, quetiapine, and ziprasidone easily preferable as first-line therapies for psychosis, with conventional antipsychotics and clozapine as second-line therapies. Some of the perceptions from longer-term experience deriving from clinical practice that differ from the early indications arising from clinical trials may be summarized as follows. First, different atypical antipsychotics can have clinically distinctive effects in different patients, unlike conventional antipsychotics, which mostly have the same clinical effects in different patients. Thus, median clinical effects in clinical trials may not be the best indicator of the range of clinical responses possible for individual patients. The inhibitors are shown here, and the atypical antipsychotics including clo zapine, quetiapine, ziprasidone, and sertindole, are shown as well. If this agent is stopped in a patient who is receiving an atypical antipsychotic that is a substrate for this same enzyme. Third, atypical antipsychotics may not work as fast as conventional antipsychotics for acutely psychotic, aggressive, agitated patients requiring sedation and onset of action within minutes; for such patients, conventional antipsychotics or sedating benzodiazepines may be useful as adjuncts or as substitutes. Finally, although virtually all studies are head-to-head comparisons of monotherapies and/or 444 Essential Psychopharmacology placebo, many patients receive two antipsychotic drugs in clinical settings. Use of Atypical Antipsychotics for Positive Symptoms of Schizophrenia and Related Disorders Although the usefulness of the atypical antipsychotics is best documented for the positive symptoms of schizophrenia, numerous studies are documenting the utility of these agents for the treatment of positive symptoms associated with several other disorders (discussed in Chapter 10; see. In fact, current treatment standards have evolved in many countries so that atypical antipsychotics have largely replaced conventional antipsychotics for the treatment of positive psychotic symptoms except in a few specific clinical situations. One area of continuing use for conventional antipsychotics is in an especially acute setting with an uncooperative patient, where a drug with not only a rapid onset of action but also an intramuscular dosage formulation may be preferred. In practice, this can mean using sedating benzodi azepines as well as a few of the old-fashioned conventional antipsychotics available for intramuscular administration, such as haloperidol and loxapine.
Phenytoin 100mg lowest price
Lower ing the urate level does not necessarily prevent the development of gout medications quizlet buy phenytoin paypal, and most of these patients will never develop any symptoms. Acute gouty arthritis is treated with therapies to reduce the inflammatory reaction to the presence of the crystals, all of which are most effective if started early in the attack. During intercritical gout, the focus shifts to preventing further attacks by lowering uric acid levels to less than 6. Dietary restriction is mainly aimed at avoid ing organ-rich foods, such as liver, and avoiding alcohol. Patients taking thiazide diuretics should be switched to another antihypertensive if possible. Urate lower ing can be accomplished by therapy to increase uric acid excretion by the kidney, such as with probenecid. Uricosuric agents such as this are ineffective in patients with renal failure, however, and are contraindicated in patients with a history of uric acid kidney stones. In these patients, allopurinol can be used to diminish uric acid production, but given at lower doses in patients with renal disease. Febuxostat is a new xanthine oxidase inhibitor that does not require dose adjustment in renal insufficiency. Patients with tophaceous gout are managed as previously described during acute attacks and subsequently treated with allopurinol to help tophaceous deposits resolve. Surgery may be indicated if the mass effect of tophi causes nerve compres sion, joint deformity, or chronic skin ulceration with resultant infection. She has mild swelling and erythema of her ankle, and pain on passive exion of her wrist. Joint aspirate reveals numerous leukocytes and polymorphonuclear leukocytes, but no organisms on Gram stain. The patient described best ts the picture of disseminated gonococcal infec tion. She has the rash, which typically is located on extensor surfaces of distal extremities. Nafcillin would be useful for staphylococcal arthritis and would be the more likely choice if she were older, had some chronic joint disease such as rheumatoid arthritis, or were immunocompromised. Gonococcal arthritis is the most common cause of infectious arthritis in patients younger than 40 years. Indomethacin or colchicine would be useful if she had a crystalline arthritis, but that is unlikely in this clinical picture. Intraarticular prednisone is contraindicated until infectious arthritis is ruled out. Synovial uid cultures usually are sterile in gonococcal arthritis (in fact, the arthritis is more likely caused by immune complex deposition than by actual joint infection), and blood cultures are positive less than 50% of the time. Diagnosis is more often made by nding gonococcal infection in a more typical site, such as urethra, cervix, or pharynx. The inam matory arthritis as shown by Gram stain of the joint aspirate is suspicious for infection, even with no organisms seen on Gram stain. In a febrile patient with a joint effusion, diagnostic arthrocentesis is man datory. Inflammatory fluid (white blood cell count more than 2000/mm3) should be considered infected until proven otherwise. Gonococcal arthritis usually presents as a migratory tenosynovitis, often involving the wrists and hands, with few vesiculopustular skin lesions. Nongonococcal septic arthritis is most often caused by S aureus and most often affects large weight-bearing joints. Monosodium urate crystals in gout are needle-shaped and negatively birefringent (yellow) under the polarizing microscope. Calcium pyro phosphate dihydrate crystals in pseudogout are rhomboid and positively birefringent (blue). Joint pain and stiffness are making it harder for her to get out of bed in the morning and are interfering with her ability to perform her duties at work. She also reports malaise and easy fatigability for the past few months, but she denies having fever, chills, skin rashes, and weight loss. Physical examination reveals a well-developed woman, with blood pressure 120/70 mm Hg, heart rate 82 bpm, and respiratory rate 14 breaths per minute. Its major distinctive feature is a chronic, symmetric, and erosive synovitis of peripheral joints, which, if untreated, leads to deformity and destruction of joints due to erosion of cartilage and bone. The presence of soft tissue swelling and tenderness with limited active range of motion but normal passive range of motion suggests the problem is extraarticular soft tissue inflammation, such as bursitis or tendonitis. If symp toms are relatively acute (<6 weeks), the major considerations are arthritis due to viral infection (such as hepatitis B or C, rubella, or parvovirus B19) or the earliest manifestation of a true rheumatic disease. Viral serologies and compatible clinical history of exposure often can make the diagnosis at this point and obviate need for further rheumatologic evaluation. Rheumatic fever, which can cause symmetric polyarthritis, is an acute febrile illness lasting only 6 to 8 weeks. The vast majority of patients have peripheral joint involvement of more than five joints. Inflammation is not limited to the joints but also occurs at the periosteum, along tendons, and at the insertion points into the bone, resulting in the development of sausage digits, which are typical of psoriatic arthritis (and Reiter syndrome). Although the arthritis can precede the development of a skin rash, the definite diagnosis of psoriatic arthritis cannot be made without the evidence of skin or nail changes typical of psoriasis (nail pitting, scaly plaques). Reiter syndrome is a form of reactive arthritis with the triad of arthritis, uveitis, and urethritis. Degenerative joint disease may affect multiple joints, but it occurs in older age groups, is usually not associated with inflammation or constitutional symptoms, and tends not to be episodic. Morning stiffness or stiffness after any prolonged inactivity is a common fea ture of many arthritic disorders. Rheumatoid nodules are subcutaneous nodules typically found over extensor sur faces of the proximal ulna or other pressure points. On x-rays, the typical findings are joint space narrowing, subchondral polysclerosis, marginal osteophyte formation, and cyst formation. Usually, though, the typical x-ray findings do not develop until later in the disease process after a diagnosis has been made based on clinical findings. The structural damage to the joint is irreversible and worsens with disease progression. Multiple different joints may be affected, such as hand, foot, ankle, hip, shoulders, elbow, and cervical spine. Although not common, the continuous bone erosion may result in an atlantoaxial subluxation with cervical dislocation and spinal cord compression. At this stage in the disease process, our patient is presenting with joint com plaints, fatigue, and malaise. Usually, the development of extraarticular phenomenon allows the physician to make a more specific diagnosis. Corticosteroids have an immediate and dramatic effect on joint symptoms, but were historically thought not to alter the natural progression of the disease. Recent evidence suggests that low-dose cortico steroids may retard the progression of bone erosions. Physical examination shows warmth and swelling of both knees with large effusions. Arthrocentesis of the right knee reveals the presence of intracellular and extracellular weakly positive birefringent crystals in the synovial uid. Physical examination shows exquisite tenderness of the joint, with swelling, warmth, and erythema. Synovial uid analysis and aspiration are most likely to show which of the following On physical exami nation, maculopapular and pustular skin lesions are noted on the trunk and extremities. He reports dif culty in getting out of bed in the morning and may have to roll out sideways, trying not to ex or rotate the spine to minimize pain.